+86 18186671616
+86 18186671616 Jason@cleanroomequips.com
Jason@cleanroomequips.com MENU
MENU
1 Scope
This standard specifies the basic requirements for the production and quality management of sterile medical devices and their components: the production of primary packaging materials for sterile medical devices shall also comply with the provisions of this standard.

2 Referenced standards
The following standards contain provisions. The provisions of this standard are constituted by reference in this standard. When this standard is published, the versions shown are valid. All standards will be revised, and parties using this standard should explore the possibility of using the latest version of the following standards.
GB/T6583--1994 Quality management and quality assurance
GB/TI6292--1996 Test method for floating particles in clean rooms (areas) of Pharmaceutical industry
GB/T16293--1996 Test method for floating bacteria in clean rooms (areas) of pharmaceutical industry
GB/T16294--1996 Test method for settling bacteria in clean rooms (areas) of pharmaceutical industry
YY/T 0313--1998 Packaging, marking, transportation and storage of medical polymer products
JGJ71--1990 Clean room construction and acceptance specifications.
3 Definitions
This standard uses the definitions of GB/T 6583 and YY/T 0313 and the following definitions.
3.1 lot
The number of a product with the same properties and quality produced when the production conditions are relatively stable.
3.2 lot number
A group of numbers or letters and numbers used to identify "lots"
3.3 production lot
Refers to the number of products with the same properties and quality produced continuously under the same process conditions over a period of time. Note: For some continuously produced products, it is sometimes difficult to divide the production batches. For management needs, the products produced on each working day or shift are often used as production batches.
3.4 sterilization lot
The number of products with the same sterility assurance level sterilized under the same process conditions in the same sterilization cabinet.
3.5 secondary sterilization
A confirmed process used to make the product free of any form of viable microorganisms.
3.6 sterile
The absence of viable microorganisms on medical devices.
3.7 primary package
Packaging in direct contact with sterile medical devices.
3.8 sterile medical device
Refers to any medical device marked "sterile".
3.9 Clean room (area)
A room (area) that needs to control dust particles and microorganisms. Its building structure, equipment and functions all have the function of reducing the intervention and retention of pollution sources in the room (area).
3.10 Cleanliness
The allowable statistical number of suspended particles greater than or equal to a certain particle size in a unit volume of air in a clean environment.
3.11 Air purification
The act of removing pollutants from the air to make the air clean.
3.12 Personnel purification room

Auxiliary room where personnel are purified according to a certain procedure before entering the clean room (area)
3.13 Material purification room

Auxiliary room where materials are purified according to a certain procedure before entering the clean room (area).
3.14 Material
Refers to raw materials, auxiliary materials, packaging materials, purchased (co-purchased) spare parts, etc.
4 Quality system
Sterile medical device manufacturers should establish and implement an effective quality management system, form a complete set of quality management system documents, and conduct management reviews and internal audits regularly. Note 1 GB T19001 and VYIT0287 or GBIT 19002 and YIT0288 stipulate the requirements of the quality management system.
4.1 Quality Policy
The quality policy should be issued by the enterprise's top management in the form of documents, and should ensure the establishment of quality goals and the understanding and implementation of the quality policy at relevant functions and levels.
4.2 Organizational Structure
The enterprise should establish an organizational structure that is consistent with the quality management system and product production requirements, define its functions and relationships (including positions and authorities) and form documents to promote effective quality management.
The top management of the enterprise should designate one or more management representatives among the management personnel and define their positions and authorities.
4.3 Personnel
The enterprise should be equipped with management personnel and technical personnel of different levels and categories who have professional knowledge, production experience, organizational ability and are familiar with the relevant national medical device supervision and management regulations that are suitable for the production of sterile medical devices. Responsible for organizing production and quality management.
The top management of the enterprise must be familiar with product production technology, have organizational skills, have certain scientific and cultural knowledge, be able to organize production according to the requirements of this standard, and be fully responsible for the implementation of this standard and product quality.
The leader in charge of sterile medical device production technology and quality management in the enterprise should have a college degree or above in this major or related majors or equivalent, have practical experience in sterile medical device production quality management, and be responsible for the implementation of this standard and product quality.
The person in charge of the production management and quality management department of the enterprise should have professional knowledge and management experience suitable for their job. Be able to make correct judgments and deal with practical problems in the production and quality management of sterile medical devices.
The person in charge of the sterile medical device production management department and the quality department shall not hold concurrent positions.
Operators and quality inspectors engaged in key positions and special processes should have a high school degree or above, have received professional technical training, have basic theoretical knowledge and practical operation skills: full-time inspectors should also undergo appropriate technical training and hold certificates before taking up their posts.
A production operator should have a cultural level suitable for his job and take up his post after professional technical training.
The enterprise should train and assess the personnel in product production technology, clean environment control, medical and health knowledge, product handling, storage, protection, etc. in accordance with the requirements of this standard, and continuously improve their business capabilities and quality awareness. And keep training records.
The supply and sales department should be equipped with personnel with professional knowledge and competent for the supply, marketing and management of sterile medical devices.

5 Production environment, facilities and layout
5.1 Factory site and factory area
Sterile medical device manufacturers must have a clean production environment. The ground, road surface and transportation in the factory area should not cause pollution to the production of sterile medical devices.
The factory site should be selected in an area with good sanitary conditions, fresh air, low atmospheric dust, low archaea concentration, no harmful gases and good natural environment.
The factory site should be far away from railways, docks, airports, major traffic arteries, factories, storage warehouses, yards that emit large amounts of dust and harmful gases, and other areas with serious air pollution, water pollution, vibration or noise interference. The distance between the clean workshop and the municipal traffic road should not be less than 50m.
The main roads in the factory area should be wide and smooth, and should be built with materials that are not easy to generate dust. 5.1.4 The factory area should be reasonably laid out. The administrative area, living area and auxiliary area shall not have adverse effects on the production area. Animal rooms and sterilization workshops should be located in secluded and safe locations, and should have corresponding safety, ventilation and sewage (toxic) facilities. Their design and construction should comply with relevant national regulations.
The fields around the production plant should meet the four requirements (no stagnant water, no weeds, no garbage, and no mosquito and fly breeding grounds). There should be no exposed land.
5.2 Production plant
The production plant is divided into general production area and clean area according to the production process and product quality requirements. The plant should be reasonably laid out according to the production process and the required air cleanliness level.
5.2.1-General production area
The general production area should be reasonably designed, with good lighting and ventilation to meet production needs.
5.2.2 Clean area
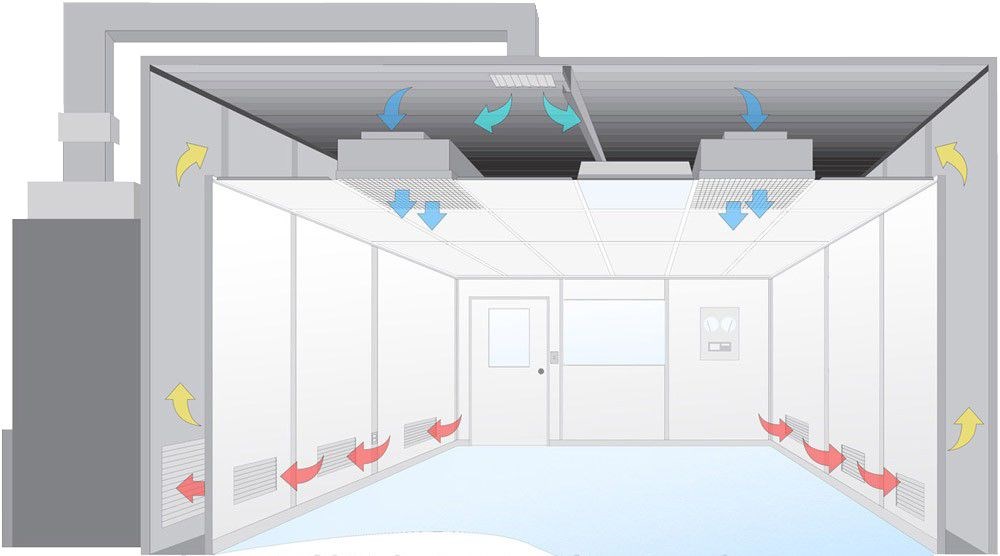
In addition to meeting 5.2.1. An air conditioning and purification system corresponding to the cleanliness level should also be configured. Appendix A gives the air cleanliness level of the clean room (area) of sterile medical devices. Appendix B gives the setting guide for the cleanliness level of the production environment of sterile medical devices.
When designing, building and decorating clean workshops, it should be easy to clean. The inner surface of the clean room (area) should be flat, smooth, without cracks, with tight joints, no grains falling off, and able to withstand cleaning and disinfection. The junction between the wall and the ground should be made into an arc or other measures should be adopted to reduce dust accumulation and facilitate cleaning. There should also be dustproof and anti-pollution facilities to prevent insects and other animals and foreign objects from mixing in.
The external windows of the personnel purification room and Clean Room (area) should be double-layer windows with good sealing. The ceiling of the clean room (area) and the pipes, air vents and walls or ceilings entering the clean room (area) should be sealed.
The door of the clean room (area) should be well sealed and open in the direction of high cleanliness.
The clean room (area) should be equipped with a safety door. It should open in the direction of safe evacuation, be well sealed at ordinary times, easy to open in an emergency, and the safety passage should be barrier-free.
The clean room (area) should be reasonably laid out according to the process flow, and the flow of people and goods should be separated and fixed in direction.
The water, electricity and other transmission lines in the clean room (area) should be concealed. The pipe openings of the electrical pipelines and the joints between various electrical equipment installed on the wall and the wall should be reliably sealed.
The clean room (area) should use lighting fixtures with simple external shapes, not easy to accumulate dust, and easy to wipe. The lighting fixtures should be installed openly rather than suspended. When using ceiling installation. Reliable sealing measures should be taken at the joints between the lamps and the ceiling.
The operating table should be smooth and flat. There should be no gaps, no dust particles and fibers falling off, no dust accumulation, easy to clean and disinfect, and wooden or painted countertops should not be used.
The compressed air and other gases used in the clean room (area) should be purified. In particular, the cleanliness of the gas that contacts the surface of the product should be verified and routinely controlled to adapt to the products produced.
The pool and floor drain in the clean room (area) shall not contaminate sterile medical devices.
5.3 Personnel purification
The personnel purification room shall include a shoe changing room, a coat storage room, a washing room, a clean work clothes wearing room, an airlock room or an air shower, etc.
Personnel entering the sterile medical device production clean room (area) or sterile operation clean room (area) should be purified. Appendix D gives the general procedures for entering and leaving the clean room (area).
At the shoe changing area entering the personnel purification area, pay attention to the two types of shoes not to cross-contaminate. There should be a clear boundary between the shoes for going out and the shoes to be changed that is not easy to cross at will. Slippers should not be worn in the clean room (area).
The flow of personnel should strictly follow the direction from the low cleanliness area to the high cleanliness area. 5.3.5 The faucet in the washroom shall be set at one for every 10 people in the maximum shift, and the faucet should not be opened and closed manually.
The entrance and exit doors of the airlock room shall be prevented from being opened at the same time. When setting up a single-person air shower, one should be set for every 30 people in the maximum shift. When the number of staff in the clean room (area) exceeds 5, a one-way bypass door shall be set on one side of the air shower.
The average area per person for staff in the clean room (area) shall not be less than 4 m2.
5.4 Material purification
Materials entering the clean production area shall have cleaning measures, such as outer packaging room, dust removal room, etc.
An airlock room or double-layer transfer window shall be set between the material purification room and the clean room (area) for transferring materials and other items.
The outer packaging for material transportation and storage and packaging materials that are easy to shed dust and fibers shall not enter the clean room (area). The primary packaging materials that directly contact the product should be able to effectively prevent contamination during transportation, storage and delivery, and at least two layers of sealed packaging.

5.5 Process layout
The clean room should be arranged according to the product formation process. The process flow should be compact and reasonable. The material transfer route should be as short as possible to facilitate operation and process control. The flow of people and logistics should be strictly separated and cross-flow is prohibited.
Clean rooms (areas) can only be equipped with necessary process equipment and facilities. There should be space suitable for the production scale to store the intermediate products or products produced in the clean room (area) and as close as possible to the production area associated with it to reduce mixing and contamination during transportation. The storage area should be arranged with waiting areas, qualified areas and unqualified areas, with obvious signs.
Clean rooms (areas) with high air cleanliness should be arranged in areas where people pass or arrive the least. Clean rooms (areas) of different cleanliness levels should be arranged from high to low from inside to outside. There should be anti-pollution measures such as airlocks or double-layer transfer windows when clean rooms (areas) of different levels are connected to each other.
When conveying materials between areas with different air cleanliness, if conveyor belts are used, in order to prevent cross-contamination, conveyor belts should not pass through partitions, but should be conveyed in sections on both sides of the partitions. For the material transfer between areas with different air cleanliness in the production area of non-sterile products, it must be conveyed in sections, unless the transfer device adopts continuous disinfection.
For instruments that need to be cleaned in a clean room (area), the air cleanliness level of the clean room should be consistent with the product requirements. Equipment and instruments in Class 100 and Class 10,000 clean rooms (areas) should be cleaned outside this area, and the air cleanliness of the cleaning room should not be lower than Class 100,000:
Cleaning tools should be washed, dried and sanitary ware stored in an independent, sanitary and well-ventilated tool room. Sanitary ware should not be stored in A Clean Room (area).
6 Equipment and tooling
6.1 The design and selection of equipment should meet production requirements, with a reasonable layout and easy operation, repair and maintenance.
6.2 The equipment and tooling used in the clean room (area) should have dust and pollution prevention measures, with simple structure, low noise and dust-free operation. The surface of equipment, tooling and pipelines should be smooth and flat, without any particulate matter falling off, easy to clean and disinfect or sterilize, and can reduce pollution.
6.3 The surface of equipment, tooling and pipelines in direct contact with materials or products should be non-toxic, corrosion-resistant, without dead corners, easy to clean and disinfect or sterilize, and not chemically react with materials or products.
6.4 Lubricants, coolants, cleaning agents used in equipment and release agents used for parts that are not cleaned after mold forming in the clean area shall not cause pollution to the product
6.5 An independent mold room (or area) should be set up for mold maintenance and storage to prevent mold contamination of the clean room (area).
6.6 A sufficient number of workstations should be equipped with good sealing and easy to clean and disinfect. The working tools in the clean room (area) and the general production area should be strictly separated and clearly marked, and shall not be cross-used.
6.7 There should be equipment for preparing process water, and the water production capacity should meet production needs; process water should be tested regularly according to standards; the storage and delivery pipelines of process water should be made of stainless steel or other non-toxic materials: they should be cleaned and disinfected regularly.
6.8 The scope of application and accuracy of instruments, meters, measuring tools, scales, etc. used for production and inspection should meet the requirements of production and quality inspection, should have obvious status identification, and be calibrated or verified according to the prescribed cycle.
6.9 Equipment and tooling should be maintained, serviced and verified regularly. When equipment is updated, it should be verified and can only be used when it is determined that there is no impact on product quality, see Appendix E.
6.10 There should be regulations for the management of production and inspection equipment (including spare parts), tooling and workstation equipment, and equipment files should be established to keep records of equipment use, maintenance, repair and improvement.
7 Procurement and material management
7.1 The enterprise should have regulations to control the procurement process, prepare procurement documents such as procurement plans, contracts, technical agreements, etc., clearly state the quality requirements of the purchased materials to ensure that they meet the standard requirements, and keep copies of the procurement documents.
7.2 The supplier should be evaluated, and its production environment (especially when there is a purification requirement), quality assurance, whether it has certificates and licenses that meet national regulations, reputation, etc. should be investigated and analyzed. A small amount of trial should be carried out before placing a large order, and it can only be used for production after passing the inspection. The supplier should be relatively stable. The supplier's quality records should be established and preserved.
7.3 After entering the factory, the purchased materials should be placed in an area with a clear "pending inspection" sign, and the warehousing procedures can be handled only after passing the inspection by the quality inspection department.
7.4 Materials should be stored in a warehouse with temperature and Relative humidity that meet their respective requirements, no corrosive gases, good ventilation, and fire protection measures. Pending inspection products, qualified products, and unqualified products should be strictly separated, with status identification, which can effectively prevent mixing. All kinds of materials are classified and stored in batches, and storage cards are written.
7.5 The distribution of materials should be recorded and signed by the person who issues and receives the materials. The distribution of materials should follow the principle of first-in-first-out.
7.6 Labels, certificates of conformity, instructions for use, and small packages should be kept by a dedicated person, and their distribution, use, and destruction should be recorded.
7.7 Special isolation measures should be taken for flammable and explosive materials.

8 Quality Management
8.1 The enterprise shall establish a quality management department under the direct leadership of the top management, and the person in charge of the quality management department shall meet the requirements of Article 4.3.4
8.2 The quality management department shall be equipped with a certain number of quality management and inspection personnel, and have physical, chemical and biological laboratories and testing equipment that are suitable for the production scale, variety and inspection requirements of sterile medical devices.
8.3 The duties and powers of the quality management department:
Responsible for the quality management and inspection of the entire production process of sterile medical devices. Formulate inspection specifications according to product standards and quality requirements:
Have the right to approve or determine the use of all materials and intermediate products and the delivery of products:
Decide whether packaging materials, labels, and instructions for use are allowed;
Evaluate whether the storage conditions of materials, intermediate products, and products are applicable:
Keep samples of products for observation and regular inspection to evaluate the quality stability of products and provide a basis for determining the shelf life of products;
Review the procedures for handling non-conforming products and the procedures for corrective and preventive measures; determine the handling methods for returned, recalled and non-conforming products;
Management of equipment, instruments, reagents and measuring instruments for inspection and testing:
monitoring and recording of clean rooms (areas) and process water.
8.4 The quality management department shall conduct incoming inspection or verification, process inspection, and final product inspection in accordance with regulations. And issue inspection records and reports; the records and/or reports shall be signed by the person responsible for executing the inspection and authorizing the release of products.
8.5 The quality management department shall conduct sampling in accordance with regulations, and the sampling shall be representative
8.6 The quality management department shall evaluate the supplier together with relevant departments.
9 Production process management
9.1 The enterprise shall control the entire production process of the product.
9.2 Before the product is officially put into production, the production process shall be fully verified to determine the feasibility of the process.
9.3 Special processes and key processes shall be set up with quality control points, and control point management documents and work instruction documents (such as process cards or work instructions, etc.) shall be formulated for continuous monitoring and control, and all control parameters shall be recorded.
9.4 According to the different requirements of the product for the cleanliness of the production environment, various operation processes are limited to designated production areas. Personnel and items entering the clean room (area) must be purified according to the purification procedures for personnel and materials required by the corresponding product. The transfer and use of work tools in different cleanliness areas should prevent cross contamination.
9.5 Intermediate products in the production process stored in the clean room (area) should have measures to prevent contamination. And there are product names and inspection status labels, and unqualified intermediate products should be stored and recorded separately to prevent mixing.
9.6 For spare parts that need to be cleaned, they should be cleaned in a clean room (area) of the corresponding level with process water of the corresponding requirements. The cleaning water and cleaning process should be confirmed and routinely controlled to adapt to the products produced.
9.7 Product identification and traceability
Manufacturers of sterile medical devices should stipulate that appropriate methods should be used to identify products throughout the production process. Each batch or each product should have a clear and firm unique identification and records during the formation process to ensure traceability.
The enterprise should formulate control documents for batch number (production batch number and sterilization batch number) management. Each batch or each product should have a quality record reflecting product identification and input materials, production process (including clean room (area) Environmental monitoring, key workstations and special processes such as sterilization parameters, etc.). Control of the use of relevant equipment, production date, signatures of operators and reviewers, and inspection results.
9.8 Packaging, marking, labeling and instructions for use
Sterile medical devices must be sealed. The packaging materials should be selected and designed according to the product performance and sterilization method, and should meet the storage requirements. Note: ISO/DIS11607 specifies the basic requirements for evaluating the performance of sterile medical device packaging.
The largest unit of sterile packaging for sterile medical devices should be a single package. The word "sterile" and/or symbol should be marked on the single package, and the words "damaged packaging is prohibited from use" should be included.
The markings on the packaging should correctly guide the transportation, storage, unpacking and use of the product, and should be obvious, clear and firm, and should not fall off or become blurred due to the sterilization, transportation and storage processes used.
The markings on the packaging must meet the requirements of the corresponding product standards.
9.9 Secondary sterilization
Sterile medical devices that need to be sterilized should use a confirmed sterilization method to sterilize enzymes to ensure the reliability of the sterilization effect. Note: For appropriate sterilization methods and confirmation and routine control requirements of the sterilization process of medical devices, see ISO 11131, ISO11135, and ISO11137.
Products before and after sterilization should be strictly separated, labeled, and strictly distinguished from qualified products.
Operators should perform sterilization operations in strict accordance with the provisions of the documents, and have complete records of the sterilization process and parameters.
9.10 Control of non-conforming products
The enterprise should formulate a procedure document for the control of non-conforming products to prevent the unintended use or delivery of non-conforming products.
Non-conforming products. Should be labeled, registered, evaluated, isolated and disposed of.
Non-conforming products can only be accepted if they meet regulatory requirements: if rework is required, the adverse effects of rework on the product should be confirmed, and re-inspection and records should be made according to regulations after rework.
9.11 Corrective and preventive measures
The enterprise should formulate and implement a procedure document for corrective and preventive measures.
The enterprise shall effectively handle customer complaints and non-conformity reports: investigate the causes of non-conformity related to the product production process and quality management system: take corrective measures and verify.
The enterprise shall use various information sources to discover and analyze potential factors of non-conformity, take preventive measures and implement control.
If the enterprise does not take corrective and preventive measures for customer complaints, it shall record the reasons.

10 Hygiene management
The enterprise shall formulate hygiene management documents that are consistent with product quality requirements and production processes, and implement them conscientiously and keep records
10.1 Clean room (area) hygiene
Clean rooms (areas) shall be regularly cleaned, washed and disinfected in accordance with the provisions of the documents, and the disinfectants or disinfection methods used shall not cause contamination to equipment, tooling, materials and products. The types of disinfectants shall be changed regularly.
Clean rooms (areas) shall be regularly monitored and recorded in accordance with the requirements of Appendix C.
10.2 Personal hygiene
The enterprise should formulate a hygiene code for operators, which should include regular haircuts, bathing, nail trimming, no makeup, no jewelry, and no personal items brought into the clean room (area), etc.: and there should be a special person to check
The enterprise should establish employee health records, and operators who directly contact materials and products should have a physical examination at least once a year. Personnel with infectious and contagious diseases are not allowed to engage in work that directly contacts products.
Personnel entering the clean room (area) must be purified according to the personnel purification procedures required by the corresponding products. And wear clean work clothes, work hats, masks, and work shoes. Operators who directly contact the product should disinfect their hands again at regular intervals.
10.3 Process hygiene
Equipment and pipelines should be cleaned regularly and kept clean, without running, bubbling, dripping, or leaking.
Parts of equipment and tooling that are in direct contact with products, as well as work surfaces and workstation tools, should be cleaned and disinfected regularly to keep them clean. The workstations and equipment in the clean room (area) should be cleaned and disinfected with purified water in the clean room (area).
Clean work clothes should be made of materials with smooth texture, no static electricity, and no shedding of fibers and granular substances. Their work clothes and hats should be able to effectively cover underwear and hair. Clean work clothes used at different cleanliness levels should be regularly cleaned, dried and sorted in the clean environment of the corresponding level.
The clean room (area) is limited to production operations in the area and approved personnel.
11 Product sales and user services
11.1 Enterprises should establish user files, frequently contact users, actively solicit user opinions, and provide services to users in a timely manner.
11.2 Each batch of products should have sales records, and once unqualified products are found, they can be recovered immediately. The records should be retained for at least one year after the expiration date.
11.3 User complaints can be handled in a timely manner, and information on the sales service process should be fed back to the relevant functional departments in a timely manner, and measures should be taken and recorded.
11.4 The enterprise shall establish a medical device adverse event reporting system and designate a special agency or personnel to be responsible for the management. Adverse events should be reported to the local drug supervision and administration department in a timely manner.
11.5 Establish a product accident reporting system and a product recovery system. When a sterile medical device has a major quality problem, it should be reported to the local drug supervision and administration department in a timely manner. If a product that has been shipped is found to be unqualified due to sample observation or national spot check, it should be recovered immediately and handled according to the unqualified product control procedure.